There is
increasing concern about the reliability of biomedical
research, with recent articles suggesting that up to 85% of research
funding is wasted. This article argues that an important reason for
this is the inappropriate use of molecular techniques, particularly in
the field of RNA biomarkers, coupled with a tendency to exaggerate the
importance of research findings.
The real-time
reverse transcription polymerase chain reaction (RT-qPCR) addresses the
evident requirement for quantitative data analysis in molecular
medicine, biotechnology, microbiology and diagnostics and has become
the method of choice for the quantification of mRNA. Although it is
often described as a ‘‘gold’’ standard, it is far from being a standard
assay. The significant problems caused by variability of RNA templates,
assay designs and protocols, as well as inappropriate data
normalization and inconsistent data analysis, are widely known but also
widely disregarded. The widespread use of this technology has resulted
in the development of numerous protocols that generate quantitative
data using:
fresh, frozen or archival FFPE
(formalin-fixed, paraffinembedded) samples,
whole-tissue biopsies, microdissected samples,
single cells, tissue culture cells,
total or mRNA,
a range of different cDNA priming strategies,
different enzymes or enzyme combinations,
assays of variable efficiency, sensitivity and
robustness,
diverse detection chemistries, reaction
conditions, thermal cyclers and
individual analysis and reporting methods.
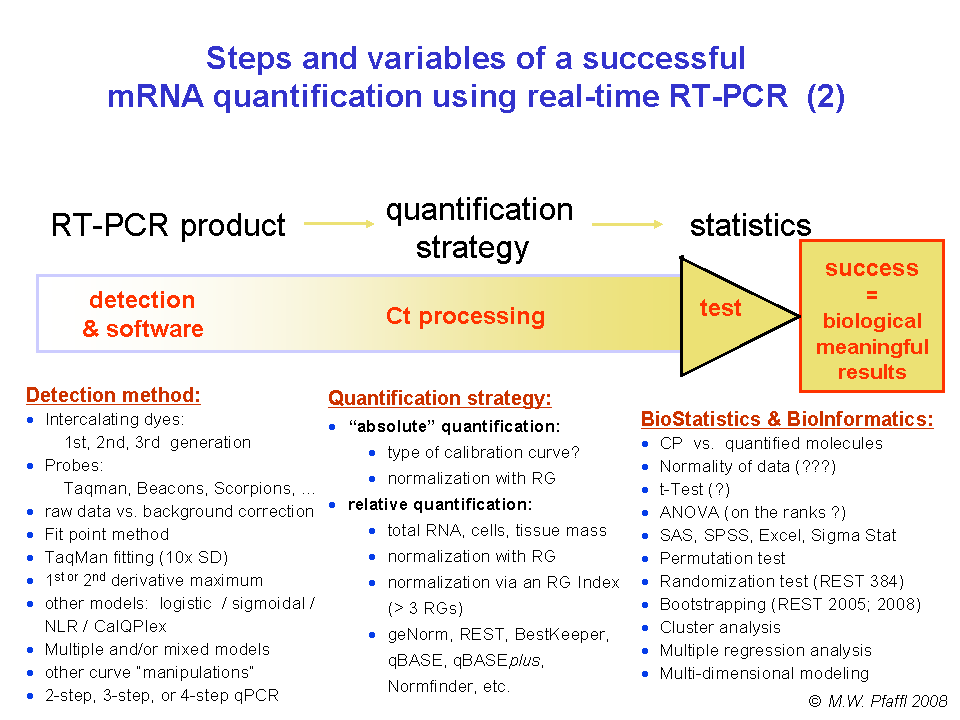
This obvious lack
of standardization at every step of the assay (Figure
1) is exacerbated by significant differences in sample
processing, use of controls, normalization methods and quality control
management and has serious implications for the reliability, relevance
and reproducibility of RT-qPCR. An overview of the considerations
relating to procedures and alternative steps for carrying out the
RT-qPCR reaction is shown in Figure
2.
PCR technology is
based on a simple principle; an enzymatic reaction that increases the
initial amount of nucleic acids. This method makes it possible to
detect specific mRNA transcripts in any biological sample. Performing
RT-PCR analysis does not only comprehend this experimental PCR step.
Following the whole workflow of a RT-PCR quantitative analysis, it
starts with the sampling step, followed by nucleic acid extraction and
stabilization, cDNA synthesis and finally the qPCR where the mRNA
quantification takes place. Problems arise when optimization of the
experimental work flow becomes necessary because of high technical
variations. The PCR reaction itself is a quite stable reaction with
reproducibility between 2-8%. Therefore the source of experimental
variances can often be found in the pre-PCR analytical steps. Usually
this is neglected and optimization is done for PCR reaction only. In
this chapter – RT-PCR optimization strategies - the whole workflow of
RT-PCR experiment will be discussed, because the identification of the
source of variability is only possible following error accumulation in
every single step. Reliable data can be created when the technical
variance caused by the experimental steps is kept as low as possible.
In this chapter many recommendations to decrease the technical variance
can be found.
PCR was invented over 25 years
ago by Kary Mullis [Saiki, 1985], for which he received the Nobel Prize
in chemistry in 1993 [Malmström, 1997]. PCR is considered to be
the innovation which allowed molecular biology to evolve to the current
level. It has become an indispensable technique in life science
research and more recently in routine human and veterinary diagnostics.
PCR has evolved over the past decades from a technically complicated
method to a simple and easy to apply method. There is a wide variety of
ready-to-use reagents available that allows those with some basic
training and who master the skill of pipetting to perform a PCR.
Enzymes and instruments have been continously engineered to speed up
the PCR process, so that a PCR can presently be performed in less than
half an hour. However, the simplicity of the method is its strength and
weakness at the same time. As it is relatively easy to generate a
result many PCR users fail to appreciate the quality control that is
required to generate reliable and meaningful results. With the more
recent use of PCR in diagnostics the call for quality control is
increasing in accredited and quality aware laboratories. An increasing
number of laboratories either elect or are required to obtain an ISO
17025 [CEN, 2005] or ISO 15189 [CEN, 2007] accreditation to guarantee
the quality of the results generated. At the same time, the research
community’s call for biologically meaningful conclusions is increasing
in parallel. In 2009 a group of leading PCR scientists published
guidelines (MIQE) [Bustin, 2009], that assist qPCR users to design a
robust qPCR experiment that leads to trustworthy and biologically
meaningful results which can be reproduced in any other laboratory. The
main variables of the (q)PCR reaction are the purity and quality of the
template DNA or cDNA, the design, purity and concentration of the
primers and probes, the concentration of the different reagents, the
type of buffer and the type of enzyme, the tubes, strips or plates and
the thermocycler used (figure 1). The vast majority of (q)PCR
optimizations are performed on the variables of DNA, primers and
template. Yet, very little attention is paid to the contribution of the
variability of tubes and thermocycler to the (q)PCR result, as they are
incorrectly considered to be constants rather than variables.
Figure 1:
Main variables of qPCR process
The goal of this
Thermocycler Calibration Guide is to illustrate which types of
thermocycler variability do exist, show the impact of thermocycler
variability on the outcome of PCRs or qPCRs, and offer practical
solutions how to eliminate or control thermocycler variability. The
practical protocols allows us to put into practical use the data from
CYCLERtest Calibration Certificates and Reports. Examples will be given
showing how thermocyclers can be aligned and programmed to mimic each
other. Furthermore, examples will be given showing how calibration
results can be used for validation purposes when working under ISO
17025, ISO 15189 accreditation and many other regulations. This guide
will allow PCR and qPCR users to explore the full potential of
CYCLERtest thermocycler calibration data.
Having problems with your gene expression or SNP genotyping
experiments? Do your amplification curves look sigmoidal, or do you
have no curves at all? Do your allelic discrimination plots have
diffuse or trailing clusters? Our interactive troubleshooting tool will
guide you step by step to a solution.
Stephen A. Bustin, Vladimir
Benes,
Jeremy A. Garson, Jan Hellemans, Jim Huggett, Mikael Kubista, Reinhold
Mueller, Tania Nolan, Michael W. Pfaffl, Gregory L. Shipley, Jo
Vandesompele, & Carl T. Wittwer
Clinical Chemistry 2009,
55(4): 611-622
BACKGROUND:
Currently, a lack of consensus exists on how best to perform and
interpret quantitative real-time PCR (qPCR) experiments. The
problem is exacerbated by a lack of sufficient experimental detail in
many publications, which impedes a reader's ability to evaluate
critically the quality of the results presented or to repeat the
experiments. CONTENT: The
Minimum Information for Publication of Quantitative Real-Time PCR
Experiments (MIQE) guidelines target the reliability of results to help
ensure the integrity of the scientific literature, promote consistency
between laboratories, and increase experimental transparency. MIQE is a
set of guidelines that describe the minimum information necessary for
evaluating qPCR experiments. Included is a checklist to accompany the
initial submission of a manuscript to the publisher. By providing all
relevant experimental conditions and assay characteristics, reviewers
can assess the validity of the protocols used. Full disclosure of all
reagents, sequences, and analysis methods is necessary to enable other
investigators to reproduce results. MIQE details should be published
either in abbreviated form or as an online supplement. SUMMARY:
Following these guidelines will encourage better experimental practice,
allowing more reliable and unequivocal interpretation of qPCR results. http://miqe.gene-quantification.info/
Nucleic
Acids Research Group (NARG)
The Nucleic Acid Research Group
(NARG)
of the Association of
Biomolecular Resource Facilities (ABRF) qPCR survey. The aim of the
survey was to determine the current status of
real-time PCR technology in laboratories around the world, particularly
core laboratories. Your answers will help us "take the pulse" of the
real-time qPCR community => http://www.abrf.org/index.cfm/group.show/NucleicAcids.32.htm
Mission Statement
To facilitate the use of nucleic acid-based
technologies in research
To examine current trends of nucleic acid-based
technologies
To assist member laboratories in self-evaluation
and growth
To promote, encourage, and reward excellence in
member laboratories
NARG 2007 Real-Time PCR Survey The
Nucleic Acid Research Group (NARG)
of the Association of Biomolecular Resource Facilities (ABRF) invites
anyone who uses “Real-Time” quantitative PCR (qPCR) to participate in
our on-line survey. The aim of the survey is to determine the current
status of real-time PCR technology in laboratories around the world,
particularly core laboratories. Your answers will help us "take the
pulse" of the real-time qPCR community. Submissions are anonymous and
results will be freely available via a "web poster". This survey will
be “open” until February 2, 2007. Results will be presented at the ABRF
2007 annual meeting in Tampa Bay, FL, Mar 31-Apr 3, 2007 and will be
available "on line" by May 1, 2007. We think it will be worth your time
to participate in this study.
- View_2007_Survey_Presentation
- View_2007_Survey_Poster
- View_2007_Survey_Questions
- View_2007_Survey_RawData
4)
NARG 2006
Study: Priming Strategies for Real-time RT-PCR The purpose of
this study is to provide an opportunity for participating laboratories
to gain crucial information about the variability of the RT-step of the
qPCR assay and about the comparability of qPCR results obtained using
different cDNA priming strategies. In addition, the study will act as
an audit for participating laboratories, who will be able to compare
the results from their protocols, techniques and equipment with those
from other laboratories around the world. The study is open for those
who use Taqman® probe-based or SYBR Green I-based assay systems.
Deadline for sample submission is Dec. 15, 2005. Data will be presented
at the ABRF 2006 annual meeting in Long Beach, CA, February 11 - 14,
2006. We think it will be worth your time to participate in this study.
- View
Study announcement (pdf) (46K)
- ViewStudy
information(pdf)
- View
Study Questions(pdf) (124K)
- View
Study Examples (xls) (68K)
- View
Slides of RG Presentation on ABRF 2006 Study (9,505K)
- View
Slides of Multiplex Development/Optimization
5)
NARG 2005
Study: Validation of Your Reverse Transcription Real-Time PCR
Technique The purpose of this study is to give investigators an
opportunity to test their reverse transcription real-time PCR technique
and to gather information about the performance of various platforms,
variations due to reagents and how people analyze their data. The study
is open for those who use both Taqman® type systems and SYBRgreen
systems. Deadline for sample submission is Dec. 15, 2004. A poster
containing a table showing (anonymously) how the individual
participants fared in their assay efforts will be posted on this page.
Data will be presented at the ABRF 2005 annual meeting in Savannah, GA,
March 5 - March 8, 2005.
- View
Study Invitation (pdf) (88K)
- View
Study Information (pdf) (96K)
- View
Submission Instructions (pdf) (54K)
- View
Poster of preliminary results
- View
Slides of Research Group Presentation
- View
Study Questions (218K)
6)
NARG 2003/2004
Real-Time PCR Survey
This survey was designed to determine the current
status of
real-time PCR technology in laboratories around the world, especially
Core laboratories. The answers allowed us to "take the pulse" of the
real-time PCR community. The survey was open from November 15, 2003
until January 15, 2004. Submissions were anonymous . Results were
presented at the ABRF 2004 annual meeting in Portland, OR, Feb 28-Mar
2, 2004 and at the Ist International qPCR symposium 3rd - 6th March,
2004 in Freising-Weihenstephan, Germany. Results are available below in
several forms. All material is copyrighted and for scientific use only.
Raw data: This is a PDF file of the raw data.
Please be aware
that there are two duplicate entries and 3 null entries in this data.
Web poster: A JPG file summarizing the survey
data.
Research Group presentation from ABRF 2004 by
Brian Holloway and Tony Yeung
NARG 2004
Taqman Primer/Probe Design Study The purpose of this study
was to give investigators an opportunity to design an optimal set of
primers/probe for a common gene and have them tested empirically for
effectiveness. We were able to demonstrate some of the basic principles
of Taqman Assay design. A table showing how the individual participants
fared in their design efforts is posted below. Final analysis of
results will be published in a peer reviewed journal. A copy of the
poster presented at the ABRF 2004 annual meeting in Portland, OR, Feb
28-Mar 2, 2004 and at the Ist International qPCR symposium 3rd - 6th
March, 2004 in Freising-Weihenstephan, Germany may be obtained by
contacting sadams@trudeauinstitute.org.
- View
study invitation (53K)
- IFNg
CDS (11K)
- Primer
3 web site
- Suggestions
for assay design/Primer 3 use (352K)
- Participant
Results (47K)
- View
Study Survey Questions (159K)
- View
Poster of preliminary results (258K)
8)
NARG 2003
Study Announcement
Real-time PCR technology is of increasing
importance in research. The
commercial cost of dual-labeled probes for real-time PCR reactions is
high because of traditional HPLC or gel purification steps. NARG
believed that these probes may be used without purification, if
carefully prepared, thereby reducing the cost and making the synthesis
of real-time PCR probes feasible and practical for any DNA synthesis
laboratory.
Reverse transcription
followed by quantitative polymerase chain reaction analysis, or
qRT-PCR, is an extremely sensitive, cost-effective method for
quantifying gene transcripts from plant cells. The availability of
nonspecific double-stranded DNA (dsDNA) binding fluorophors, such as
SYBR Green, and 384-well-plate real-time PCR machines that can measure
fluorescence at the end of each PCR cycle make it possible to perform
qRT-PCR on hundreds of genes or treatments in parallel. This has
facilitated the comparative analysis of all members of large gene
families, such as transcription factor genes (Czechowski et al., 2004).
Given the relatively low cost of PCR reagents, and the precision,
sensitivity, flexibility, and scalability of qRT-PCR, it is little
wonder that thousands of research labs around the world have embraced
it as the method of choice for measuring transcript levels. However,
despite its popularity, we continue to see systematic errors in the
application of methods for qRT-PCR analysis, which can compromise the
interpretation of results. The letter to the editor by Gutierrez et al.
in this issue highlights one of many common sources of error, namely,
the inappropriate choice of reference genes for normalizing transcript
levels of test genes prior to comparative analysis of different
biological samples. The following are 11 golden rules of qRT-PCR that,
when observed, should ensure reproducible and accurate measurements of
transcript abundance in plant and other cells. These rules are for
relative quantification of RNA using two-step RT-PCR (where the product
of a single RT reaction is used as template in multiple PCR reactions),
SYBR Green to detect gene-specific PCR products, and reference genes
for normalizing transcript levels of test genes before comparing
samples. Further details can be found elsewhere (Czechowski et al.,
2004, 2005). Most of these rules also apply to relative quantification
methods that employ sequence-specific fluorescent probes, such as
TaqMan probes, and to absolute
quantification methods.
Harvest material
from at least three biological replicates to
facilitate statistical analysis of data, freeze immediately in liquid
nitrogen, and store at –80°C to preserve full-length RNA.
Use an RNA isolation procedure that produces high-quality total RNA from all
samples to be analyzed. Check RNA quality
using an Agilent 2100 Bioanalyzer (RNA integrity number, RIN > 7 and
ideally > 9) or by electrophoretic separation on a high-resolution
agarose gel (look for sharp ethidium bromide–stained rRNA bands) and
spectrophotometry (A260/A280 > 1.8 and A260/A230 > 2.0). Quantify
RNA using A260 values.
Digest purified
RNA with DNase I to remove contaminating genomic
DNA, which can act as template during PCR and lead to spurious results.
Subsequently, perform PCR on the treated RNA, using gene-specific
primers, to confirm absence of genomic DNA.
Perform RT reactions with a
robust reverse transcriptase with no
RNaseH activity (like SuperScriptIII from Invitrogen or
ArrayScript
from Ambion) to maximize cDNA length and yield. Use ultraclean
oligo(dT) primer of high integrity. qRT-PCR gene expression
measurements are comparable only when the same priming strategy and
reaction conditions are used in all experiments and reactions contain
the same total amount of RNA (Ståhlberg et al., 2004).
Test cDNA yield
and quality. Perform qPCR on an aliquot of cDNA
from each sample, using primers to one or more reference genes that are
known to be stably expressed in the organ(s) / tissue(s) under the
range of experimental conditions tested. Threshold cycle (Ct) values
should be within the range mean ±1 for each reference gene
across all samples to ensure similar cDNA yield from each RT reaction.
Quality of cDNA can be assessed using two pairs of primers for a
reference gene that are ~1 kb apart. Typically, the Ct value for the
primer pair at the 5'-end of a cDNA will be higher than the Ct value of
the primer pair at the 3'-end, as reverse transcription begins at the
3' [poly(A)] end of the template mRNA and does not always extend to the
5'-end of the template. Ideally, the Ct value of the 5'-end primer pair
should not exceed that of the 3'-end pair by more than one cycle number.
Design
gene-specific PCR primers using a standard
set of design
criteria (e.g., primer Tm = 60 ± 1°C, length 18 to 25
bases,
GC content between 40 and 60%), which generate a unique, short PCR
product (between 60 and 150 bp) of the expected length and sequence
from a complex cDNA sample in preliminary tests, to facilitate
multiparallel qPCR using a standard PCR program. The 3'-untranslated
region is a good target for primer design because it is generally more
unique than coding sequence and closer to the RT start site.
Reduce technical
errors in PCR reaction setup by standardizing (robotize if
possible) and minimizing the number of
pipetting steps.
Mix cDNA with qPCR reagents, then aliquot a standard volume of this
"master mix" into each reaction well containing a standard volume of
specific primers. Set up reactions in a clean environment free of dust,
preferably under a positive airflow hood. Routinely check for DNA
contamination of primer and reagent stocks by performing PCR reactions
on no template (water) controls.
For relative quantification of transcript
levels, design and test
gene-specific primers for at least four potential reference genes
selected from the literature (e.g., Czechowski et al., 2005) or from
your own experience that are likely to be stably expressed throughout
all organs and treatments to be compared. Validate reference genes in
preliminary experiments on the range of tissues and treatments you wish
to compare using a foreign cRNA added to each RNA sample prior to
RT-PCR to normalize data for reference gene transcripts prior to
assessment of their expression stability (Czechowski et al., 2005).
Perform real-time
PCR on test and reference genes in parallel for
each sample to capture fluorescence data on dsDNA after each cycle of
amplification. Also, perform dsDNA melting curve analysis at the end of
the PCR run. When relying on nonspecific DNA binding fluorophors, such
as SYBR Green, to quantify relative dsDNA amount, ensure that only a
single PCR amplicon of the expected length and melting temperature is
produced using gel electrophoresis and PCR amplicon melting curve data,
respectively. We typically use a commercial mixture of hot-start Taq
polymerase, SYBR Green, and other reagents, such as Power SYBR Green
Master Mix from Applied Biosystems, and have observed significant
differences in the efficacy (PCR efficiency, specificity, and/or yield)
of such products from different suppliers.
Determine
which reference gene(s) is best for normalization of
test gene transcript levels amongst all samples (e.g., using geNorm
[Vandesompele et al., 2002] or BestKeeper
software
[Pfaffl et al., 2004]), which use as input not only the Ct value, but
also the PCR efficiency for each reaction. PCR efficiency can be
derived conveniently from amplification plots using the program
LinRegPCR (Ramakers et al., 2003). Estimation via the classical
calibration dilution curve and slope calculation is also possible,
albeit more complicated.
Finally,
calculate relative transcript abundance for each gene in each sample
using a formula that incorporates PCR efficiency for the test gene
and
Ct values for both test and reference genes.
A variety
of PCR additives and enhancing agents have been used to
increase the yield, specificity and consistency of PCR reactions.
Whilst these additives may have beneficial effects on some
amplifications it is impossible to predict which agents will be useful
in a particular context and therefore they must be empirically tested
for each combination of template and primers. Some of the more popular
of these additives are listed in the table below along with
references describing their use. http://www.staff.uni-mainz.de/lieb/additiva.html
Biochemistry32: 137 BioTechniques21: 1102 Genome Research6: 633 Nucleic Acids Research25: 3957 Proceedings of the NationalAcademy of Sciences of the United States of America70: 298 Trends in Biochemical Science22: 225
Formamide
Nucleic
Acids Research18: 7465
Non-ionic
detergents
e.g. Triton X-100, Tween 20 or Nonidet P-40 (NP-40)
Applied
and environmental microbiology62:1102-1106 BioTechniques23:504 BioTechniques25:564 Nucleic Acids Research16: 9775
T4 gene 32 protein
Applied
and Environmental Microbiology62:1102-1106
DMSO at 2-10% may be necessary for amplification
of some templates, however 10% DMSO can reduce Taq polymerase
activity by up to 50% (Gelfand 1989) so it should not be used
routinely. DMSO is thought to reduce secondary structure and is
particularly useful for GC rich templates.
A number of PCR additives are now comercially
available, however the identities of these agents are not usually
revealed by their suppliers. Frackman et al.(1998) have demonstrated (using NMR analysis) that the
PCR additive provided by QIAGEN in their PCR core kit (Q-Solution) and
that provided by CLONTECH in the Advantage-GC cDNA PCR kit is in fact
Betaine which is available at a fraction of the cost as a 5M solution
from Sigma-Aldrich (cat. # B 0300), but be sure to use Betaine or
Betaine (mono)hydrate and not Betaine HCl. Other products
suspected of consisting largely of Betaine include the "GC-RICH
solution enhancer" from Roche, "TaqMaster enhancer" from Eppendorf,
"GC-melt" from Clontech and "FailSafe enhancer" (formerly "MasterAmp
PCR Enhancment Technology") from Epicentre (Weissensteiner, pers.
comm.). Betaine is generally used at a final concentration of 1.0-1.7M.
Formamide is generally used at 1-5% and 10%
formamide is reported (Gelfand 1989) to have no effect on the activity
of Taq polymerase, however, Sarkar et al. (1990) (see table for
ref.) found that 1.25% formamide worked as well as 2.5% and 5%, and no
amplification was seen at 10% so it seems prudent not to use
concentrations of formamide greater than strictly necessary for optimal
amplification.
Non-ionic detergents stabilise Taq
polymerase and may also supress the formation of secondary structure.
0.1-1% Triton X-100, Tween 20 or NP-40 may increase yield but may also
increase non-specific amplification. As little as 0.01% SDS
contamination of the template DNA (left-over from the extraction
procedure) can inhibit PCR by reducing Taq polymerase activity
to as low as 10%, however, inclusion of 0.5% Tween-20 or -40 will
effectively neutralise this effect (Gelfand 1989).
TMAC is generally used at a final concentration
of 15-100mM to eliminate non-specific priming. TMAC has is also used to
reduce potential DNA-RNA mismatch (Proceedings of the National
Academy of Sciences of the United States of America82:
1585) and improve the stringency of hybridization reactions (Nucleic
Acids Research16: 4637).
The base analogue 7-deaza-2'-deoxyguanosine may
facilitate amplification of templates with stable secondary structures
when used in place of dGTP in a ratio of 3: 1,
7-deaza-2'-deoxyguanosine: dGTP.
BSA has proven particularly useful when
attempting to amplify ancient DNA or templates which contain PCR
inhibitors such as melanin.
REFERENCES
Frackman, S., Kobs, G., Simpson, D. and Storts, D. 1998. Betaine and DMSO: enhancing agents for PCR. Promega Notes 65: 27.
Gelfand, D. H. 1988. In Erlich, H. A.
(ed.) PCR Technology.p.17. Stockton Press, NY.
A new PT
scheme has been
developed,
providing an opportunity for users to obtain confidential and unbiased
assessment of their QPCR performance.The
PT has been developed under the DTI-funded
Measurements for Biotechnology
(MfB) programme as part of an initiative to improve the comparability
of
results between laboratories, and the materials for the first round
will be
sent out this September. The scheme takes an innovative approach,
and uses synthetic DNA targets in a proprietary artificial matrix to
allow
researchers and analysts from all sectors to participate without fears
of
laboratory contamination.Participants
will be required to perform basic DNA quantification of a high
concentration
DNA stock, DNA extraction of 9 unknown samples and QPCR analysis of the
9
unknown samples plus 3 additional unknowns. Results will be compared
between
participating laboratories, and performance will be scored using a
conventional
PT Z-scoring approach. By taking part in the scheme, participants will
be able
to demonstrate the overall effectiveness of their performance, which is
increasingly important to maintain the confidence of customers and
funding
bodies. UNIQ-PT-scheme-flier.pdf